




Here, you won’t just take in knowledge. You’ll create it.

-
20+ Degree Programs
We prepare visionary leaders, combining comprehensive excellence and collaboration across engineering disciplines.
-
10 Academic Departments
Blurring the lines between education and research, our academic programs combine theory with practical experience to address real-world problems.
-
230+ Centers, Institutes & Labs
Get hands-on experience through our interdisciplinary centers and institutes and labs, backed by the extensive resources and global connections of Johns Hopkins University.

Seeking Bold, Interdisciplinary Leaders Redefining Their Fields
A new Data Science and AI Institute will bring together experts from a wide range of disciplines to capitalize on the rapidly emerging potential of data to fuel discovery across the university.
Experience Hopkins Engineering
Hopkins Engineers are addressing today’s universal societal challenges to ensure a better tomorrow.
Realizing the Power and Promise of AI - Hopkins engineers are using AI to extend human capabilities, ensure equity in healthcare, enable a new age of space exploration, and more.

Part-time and online graduate education - Johns Hopkins top-ranked Engineering for Professionals program delivers challenging part-time, online courses in more than 22 disciplines that address the most current engineering technologies,

In the Magazine: Material Matters - Inside the historic Stieff Silver Building, Johns Hopkins has built the world’s top facility for studying the atomic structures of materials. Researchers across the Whiting School are using it to reshape fields from energy to oncology.

In the Magazine: Innovation at the Crossroads - Biomedical engineer Jennifer Elisseeff is known for asking bold questions and pursuing seemingly “outlandish” ideas that pay off big. Her latest cross-disciplinary pursuit? Unlocking the mysteries of aging.
-


Students develop wetlands plan to protect Turner Station community from rising sea level.
-
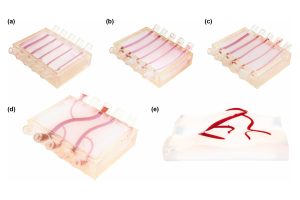
Prototype could lead to advanced, non-invasive sensors to monitor disease biomarkers.
-

Tak Igusa, professor of civil and systems engineering explains how the research team modeled projections of future casualties from trauma, infectious diseases, and other factors in Gaza.
“Hopkins has an altruistic focus that pervades all through the university … everything at Hopkins is very human-centric and people were very front-and-center. It was mission-focused. ”
 Carol Reiley, MS '07
Co-founder and former president of Drive.ai; named to Forbes’ Top 50 Women in Tech in 2018
Carol Reiley, MS '07
Co-founder and former president of Drive.ai; named to Forbes’ Top 50 Women in Tech in 2018

Pava Center
Serving first-year students to post-docs from across Johns Hopkins, Pava Center offers student entrepreneurs mentorship, programming, space, and funding support.